From Bioethics Briefings
Clinical Trials
Highlights
- Clinical trials are specifically designed to test the safety and efficacy of interventions in humans and are preceded by laboratory and animal research.
- Only about one out of every five new drugs that enters clinical testing receives Food and Drug Administration approval.
- A randomized controlled trial is a type of clinical trial comparing two or more interventions. RCTs are the principal method for demonstrating the safety and efficacy of new interventions.
- There are ethical concerns about clinical trials because some individuals are asked to accept risk in order to develop knowledge that may not directly benefit them.
- According to U.S. regulations, clinical trials must be approved by an institutional review board to determine whether they are ethical and participants’ rights are protected.
- Federal regulations also usually require that the participants give informed consent—a goal often imperfectly realized.
Framing the Issue
Clinical research with human participants utilizes a systematic approach to help understand human health and illness in order to find safe and effective ways to prevent, diagnose, and treat disease. Most clinical research in the United States is sponsored by pharmaceutical or biotechnology companies, which spent more than $10 billion on it in 2002; the U.S. National Institutes of Health is the second largest sponsor. Clinical research has a long history and has resulted in significant benefits for society, yet it continues to pose profound ethical questions.
Clinical research involves clinical trials, which are designed to test the safety and efficacy of interventions in humans. Carefully conducted clinical trials of treatments, prevention modalities, medical devices, and other interventions are considered the fastest, safest, and best way to determine whether they work for cancer, HIV/AIDS, asthma, and many other diseases. Treatment trials test experimental treatments, new combinations of drugs, or new approaches to surgery or radiation therapy. Prevention trials test medicines, vaccines, vitamins, or lifestyle changes to see if they can prevent disease or disease recurrence. There are ethical concerns about clinical trials because some individuals are asked to accept a burden or risk in order for researchers to develop knowledge that will benefit others.
Steps in Clinical Research
Clinical research follows a standard trajectory of different steps. These include:
Preclinical research
- Laboratory. Preclinical research begins in the laboratory, where researchers identify promising human interventions. Scientists often evaluate hundreds of thousands of molecules in order to find a few that have the potential to become a safe and effective treatment.
- Animal studies. The few promising therapies discovered in laboratory research are then tested in animal models to evaluate safety and desired therapeutic effect. Animal testing, although somewhat controversial, is a critical step in the process. However, the way a potential treatment works in animals may not mimic what will happen in humans. According to CenterWatch, only about one out of every 50 drugs tested in animals is determined to be safe and effective enough to test in humans.
Clinical trials
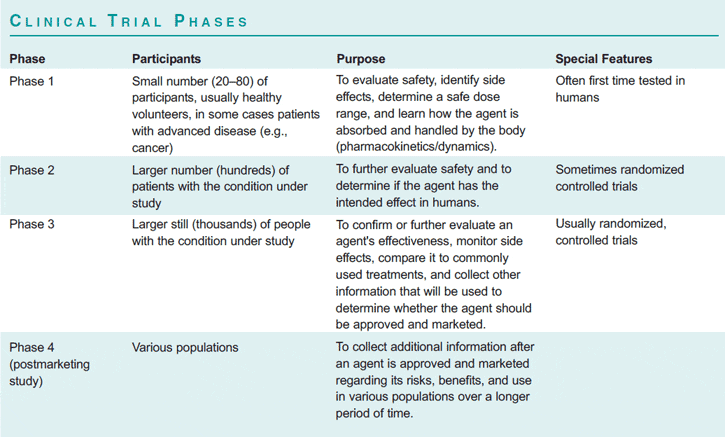
Researchers send an Investigational New Drug (IND) application to the Food and Drug Administration for permission to begin clinical trials in humans. Experimental drugs, biologics, devices, and other interventions are tested in human clinical trials to determine if they are safe and effective; what dosage is best; what the side effects are; and whether the experimental intervention is as effective as or more effective than other available interventions. A clinical trial follows a careful plan described in a written protocol that usually includes the background and purpose of the study; inclusion and exclusion criteria for participants; the schedule of tests, procedures, and other activities; the length of the study; the primary and secondary outcomes and how they will be measured; statistical methods; anticipated risks and benefits; and other details. Clinical trials are conducted in a series of stepwise phases, usually leading to a randomized controlled trial, or “RCT.” RCTs are considered by many to be the gold standard for clinical research in humans (see box, “Clinical Trial Phases”) Approximately one out of every five new drugs that enters clinical trials ultimately receives FDA approval. It can take as long as 20 years and an estimated $800 million to bring a single new drug treatment from its initial discovery through to the market.
Special Features and Challenges of Randomized Controlled Trials
A randomized controlled trial is the principal method for demonstrating the safety and efficacy of new interventions in humans. The ethical justification to begin an RCT is a lack of convincing evidence that one of two or more interventions is superior in its therapeutic efficacy, safety, or clinical usefulness. This situation is often referred to as “clinical equipoise.” An RCT aims to disturb equipoise by comparing two or more interventions to determine whether one is equivalent or superior to the others. For example, an RCT might show that individuals with cancer receiving one chemotherapy regimen live longer than those receiving a different regimen; an RCT of an HIV vaccine might aim to determine if the group of individuals receiving the vaccine have lower rates of infection than those receiving a placebo.
An RCT has several distinct features that aim to ensure scientific rigor. These include:
- Randomization. Participants do not choose, but are assigned—or randomized—by chance to either the “experimental” or “control” intervention in order to keep the two groups similar in relevant and otherwise uncontrollable aspects.
- Control. The control group is given either a standard intervention for the disease under study or a placebo.
- Blinding or masking. To reduce potential bias, RCTs are often blinded or masked. This means that neither the participant (single blind) nor the research team (double blind) knows which intervention the participant is receiving.
- Statistical evidence. Concluding that the experimental agent is better than or equal to the control agent is based on a predetermined statistical algorithm showing that the results are significant and highly unlikely to be due to chance. The commonly accepted level for statistical significance in RCTs is p £ 0.05. This is understood to mean that the probability that the relationship of variables is due to chance is less than or equal to 5%.
Ethical Considerations in Clinical Trials
Clinical trials are necessary to find out what is safe and what works so that health professionals know how to prevent and treat illness effectively. Nonetheless, there are ethical concerns about clinical trials because human research participants are a means to developing knowledge that will benefit others. Although participants may be among the beneficiaries of research knowledge, they do not necessarily benefit directly from participation in research—and, more importantly, their benefit is not the goal. The ethical principle of respect for persons requires that individuals be treated with respect for their dignity and not used merely as means for others’ ends.
Ethical codes and guidance help delimit when and how research should be conducted with human participants. The Nuremberg Code, the Declaration of Helsinki, the Belmont Report, and the U.S. Code of Federal Regulations (45 CFR 46 and 21 CFR 50, 56, and others) provide guidance for researchers to respect and protect the rights and welfare of human research participants. Most of these codes and regulations were formulated in response to historical examples of abuse, such as experimentation by Nazi doctors, the Public Health Service Tuskegee syphilis study, and others. A synthesis of this guidance and the literature suggests that to be ethical, clinical research should satisfy several criteria (see box, “Requirements of Ethical Clinical Research”).
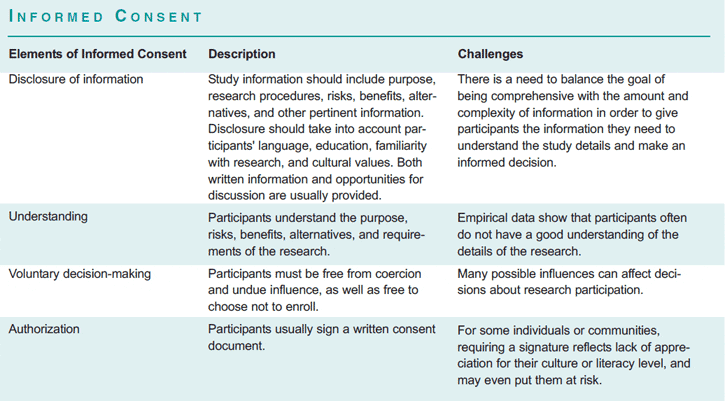
According to U.S. federal regulations, clinical trials must be reviewed and approved by an institutional review board, or IRB, before they begin and then periodically throughout the study. An IRB is a committee of physician-investigators, statisticians, community advocates, and others that determines whether a clinical trial is ethically acceptable and whether the rights of participants are adequately protected. Federal regulations also require that the participants give their informed consent, except in particular cases allowed by the regulations and deemed acceptable by an IRB—for example, for certain kinds of minimal risk research (45 CFR 46.116(d)), and some cases of emergency research (21 CFR 50.24). Informed consent is a process that involves disclosing study information to the participant so that he or she has sufficient knowledge to make an informed and voluntary decision to participate or continue to participate in the research. Although widely accepted as an integral part of ethical clinical research, the goal of well-informed individuals making voluntary choices about research participation is often imperfectly realized (see box, “Informed Consent”).

Christine Grady, RN, PhD, a Hastings Center Fellow, heads the Section on Human Subjects, Department of Bioethics, at the Clinical Center of the National Institutes of Health.
Christine Grady, “Clinical Trials,” in From Birth to Death and Bench to Clinic: The Hastings Center Bioethics Briefing Book for Journalists, Policymakers, and Campaigns, ed. Mary Crowley (Garrison, NY: The Hastings Center, 2008), 21-24.
45% of our work is supported by individual donors like you. Support our work.
Resources
- Office for Human Research Protections. Includes policy guidance, educational and public outreach material, news, and a FAQ.
- https://clinicaltrials.gov – Maintained by the National Institutes of Health. A registry of federally and privately supported clinical trials conducted in the United States and around the world that also includes resources, news, and a glossary.
- Steven Joffe and Franklin G. Miller, “Bench to Bedside: Mapping the Moral Terrain of Clinical Research,” Hastings Center Report, March-April 2008.
- Susan Gilbert, “Trials and Tribulations,” Hastings Center Report, March-April 2008.
- Karen J. Maschke, “Human Research Protections: Time for Regulatory Reform?” Hastings Center Report, March-April 2008.
- Timothy Caulfield, “Legal and Ethical Issues Associated with Patient Recruitment in Clinical Trials: The Case of Competitive Enrollment,” Health Law Review, issue nos. 2-3, 2005.
- Trudo Lemmens, “Piercing the Veil of Corporate Secrecy about Clinical Trials,” Hastings Center Report, September-October 2004.
- Hastings Center Resources on Clinical Trials
Experts
- Christine Grady, RN, PhD
